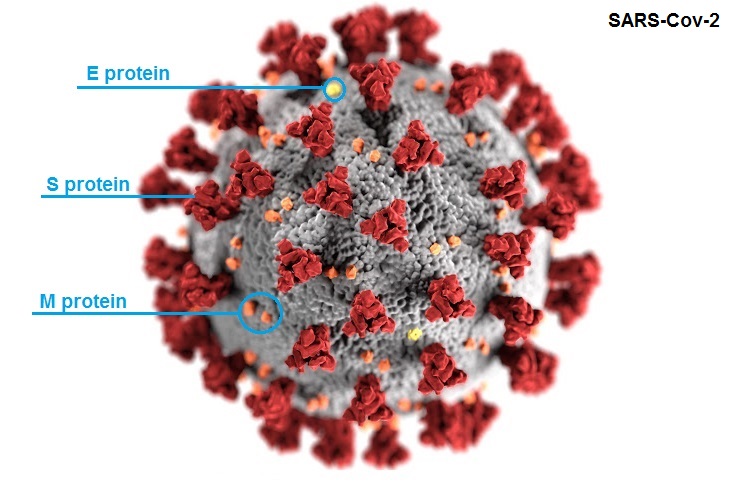
Es importante conocer la caracterización genómica y genética del coronavirus SARS-CoV2. Se trata de un virus cuyo material genético es RNA, y está compuesto por cerca de 30 mil nucleótidos, es decir, los elementos de la información genética que permiten la codificación de las diferentes proteínas de las cuales está compuesto.
El virus codifica esas proteínas, entre las cuales está su envoltura, espícula o nucleocápside del virus; todas estas proteínas están formadas por aminoácidos. Cuando cambia un aminoácido estamos hablando de una sustitución y concretamente de una mutación.
Esto ocurre con frecuencia en virus que tienen características como estas del RNA. Justamente, a lo largo de esos 30 mil nucleótidos, pueden ocurrir estos eventos que les llamaremos cambios o mutaciones.
A esta caracterización genética y a descifrar cuál es el orden de estos nucleótidos y de esos aminoácidos, se les llama secuenciación. Esta caracterización genética obtenida por secuenciación ha permitido realizar diferentes clasificaciones de este virus.
La Gisaid, una plataforma donde se depositan estas secuencias, les denomina clados y se han determinado al menos siete importantes en todo el mundo denominados S,L,V,G,GH,GR,GV para su clasificación.
Esto es de acuerdo con las características que tiene este virus genéticamente hablando, o sea, cuando va cambiando, cuando va mutando se va agrupando en diferentes clados y también se puede llevar a cabo la determinación de linajes, A, B, B.2, B.1.
En México se presentó en febrero de este año el primer caso de una variante detectada en el estado de Tamaulipas denominada variante o linaje B.1.1.7 y puede haber tantas variantes y tantos linajes de acuerdo a como este virus haya ido cambiando. Cada clado, o cada grupo genético, tiene mutaciones características que lo definen y hace que se agrupen.
Este último proceso lo hacen por medio de unas herramientas bioinformáticas que se llaman árboles filogenéticos. Estos árboles filogenéticos agrupan a estos clados, estos linajes, de acuerdo con esa asociación entre los nucleótidos. Mientras más se parezcan, mejor se agrupan entre sí, pero hay diferencias tanto en nucleótidos como en aminoácidos, pero se puede clasificar a nuestro virus SARS-CoV-2 en diferentes grupos y clados. Este es el contexto en que los científicos y especialistas mexicanos trabajan en cuanto la vigilancia de las diferentes variantes que se han detectado en México.
Recuerden que el 20 de diciembre la OMS comunicó la existencia de una variante que le denominaron en ese momento la inglesa porque se descubrió en ese país y tenía ciertas mutaciones a lo largo de su genoma que la hacían característica, entre ellas una dispersión a mayor velocidad que llamó la atención. La variante había tenido 23 mutaciones a lo largo de su genoma, y esas variaciones estaban en la proteína espícula, concretamente en el gen S.
Lo primero que hizo México fue secuenciar el fragmento en la espícula y observar si estas muestras podían o no tener esas mutaciones. El día 10 de enero se terminó de secuenciar la primera muestra de una persona inglesa que venía de un viaje y se encontró la la mutación. Se hizo la secuencia de genoma completo y detectaron que el paciente tenía las 23 variantes de dicho genoma.
El 13 de enero se empezó a trabajar en la Red Nacional de Laboratorios de Salud Pública y a emitir comunicados sobre viajeros que regresaban del exterior porque podía ser una variante importada, y en las muestras de Tampico se encontró una segunda variante, se secuenció el genoma completo y se determinó que esta mutación puede ser importada y se transmitió lateralmente entre los contactos y presentan específicamente las 23 mutaciones de la variante inglesa. Estos fueron los dos primeros casos que los denominaron caso Tampico y caso Nuevo León, porque venir de esos lugares.
Para que pueda existir o clasificar esta variante tienen que estar todas las mutaciones que son características de la misma; de hecho, se acordó por medio la OPS, de la OMS definir lo que es variante como la acumulación de cambios o de mutaciones en un genoma de un virus que lo hace diferente al original, pero con características que la hagan diferente, que tengan algún tipo de impacto.
Un impacto en salud pública es, por ejemplo, una mayor transmisibilidad, es decir, que las mutaciones estén asociadas a mayor transmisibilidad o que tenga otra asociación. Dee tal manera se determinó que lo de Jalisco es una mutación, no una nueva cepa, identificada con el código 484K
El hecho de que encontremos un cambio no nos va a indicar que tenemos una variante, tenemos que tener las mutaciones características de la variante, lo cual no ha sucedido con la mutación jalisco.
La movilización se debe a que el 27 de enero se detectaron en Jalisco cuatro casos con algunas diferencias al virus que circula en el país y surgió la duda de si se trataba de una variante o una mutación, indicó el infectólogo y exdirector de los Hospitales Civiles de Guadalajara, Héctor Raúl Pérez Gómez. De allí que se hable de mutación jalisco porque no se puede hablar de variante o nueva cepa hasta concluir la secuenciación del genoma completo.
Estimó que el hallazgo es una mutación conocida como E484K y da a lugar en una pequeña partícula del virus, un cambio en su estructura que le puede permitir, y de hecho así se ha encontrado, una mayor transmisión de la enfermedad.
La atención se centra en esta última posibilidad pues se cree que la mutación E484K está asociada con la respuesta inmunológica, es decir, evita que la persona adquiera una protección con vacunas o por haberse infectado. Y en este caso, la infección se propagaría con más fuerza.
Son ya varios instituto lo que han estado haciendo esta caracterización genética y hay una plataforma que se llama Gisaid, que es donde se depositan las secuencias genómicas que cada instituto, cada escuela, universidad, laboratorio empieza a generar, es un repositorio de todas las secuencias donde se analizan, donde se comparten las secuencias con las secuencias de Sudamérica, de Norteamérica, de Europa, de Asia y así es como se van realizando esos estudios filogenómicos para poder determinar si existen, si no existen variantes, a quién se parece, y a quién no se parece.
Se han realizado hasta el cierre de esta edición 750 secuencias de genomas completos. Estos son los institutos donde se han realizado, en el Indre por supuesto en el INER, en el IMG, en la Universidad Autónoma de Nuevo León, en Nutrición, en el IMSS, todas estas secuencias se han depositado.
La movilización se debe a que el 27 de enero se detectaron en Jalisco cuatro casos con algunas diferencias al virus que circula en el país y surgió la duda de si se trataba de una variante o una mutación, indicó el infectólogo y exdirector de los Hospitales Civiles de Guadalajara, Héctor Raúl Pérez Gómez. De allí que se hable de mutación jalisco porque no se puede hablar de variante o nueva cepa hasta concluir la secuenciación del genoma completo.
Estimó que el hallazgo es una mutación conocida como E484K y da a lugar en una pequeña partícula del virus, un cambio en su estructura que le puede permitir, y de hecho así se ha encontrado, una mayor transmisión de la enfermedad.
La atención se centra en esta última posibilidad pues se cree que la mutación E484K está asociada con la respuesta inmunológica, es decir, evita que la persona adquiera una protección con vacunas o por haberse infectado. Y en este caso, la infección se propagaría con más fuerza.
Observaciones del doctor Ernesto Ramírez González, titular de la unidad de desarrollo tecnológico e investigación molecular del Instituto de Diagnóstico y Referencia Epidemiológica (Indre) de México.